Designer ligands have promoted the first alcohol-directed activation of unreactive C-H bonds. Incorporating hydrogen-bonding interactions into the design strengthened and organised the reactive complex, an approach which could be applied to other catalytic systems.
C-H functionalisation is one of the most versatile reactions in organic synthesis but the ubiquity and inertness of these bonds make selective transformations incredibly challenging. Directed C-H activation attempts to overcome this by employing existing functionality within the substrate to anchor the metal catalyst close to a particular bond and carefully designed ligands allow chemists to use carbonyl and nitrogen-containing groups to direct the initial metal association interaction.
But alcohols – the most common functional group in nature – have proven more challenging. ‘The interaction between neutral hydroxyls and palladium is known to be weak due to a mismatch between the soft metal centre and the hard ligand. Hydroxyl groups also have more rotatable bonds than most commonly used directing groups, making the complexes more flexible,’ explains Daniel Strassfeld, a postdoc in the Yu group at The Scripps Research Institute, US. ‘Both of these mean that hydroxyl directing groups simply aren’t very good at directing palladium to the C-H bond we want to activate.’
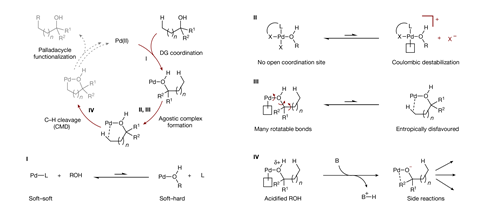
However, Strassfeld and fellow researcher Chia-Yu Chen hypothesised that by designing a ligand that promotes hydrogen-bonding, they could strengthen and organise the crucial interaction between the alcohol substrate and palladium catalyst. Using an arylation reaction as a model, they began by systematically screening bidentate ligands containing both a hydrogen-bond acceptor and an internal base.
As the palladium weakly coordinates to the hydroxyl oxygen, the hydrogen-bond acceptor interacts with the hydroxyl proton, strengthening the binding of the directing group to the catalyst. At the same time, this two-point contact between the substrate and the catalyst restricts bond rotation and holds the complex in a orientation ideal for reaction. The ligand’s internal base simultaneously forms a hydrogen bond with the hydrogen of the target C-H bond, reducing the energy barrier for the activation step and providing a third point of contact between the catalyst and substrate.
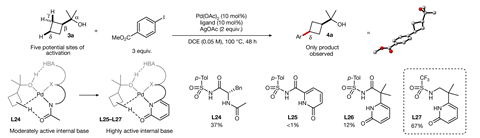
Strassfeld and Chen quickly identified promising ligand candidates for a range of benzylic and cyclobutyl substrates and next sought to validate the hydrogen-bonding mechanism using a combination of crystallographic, reactivity and computational studies. Removal of hydrogen-bond acceptor or donor groups in either the ligand or substrate resulted in no reaction whilst DFT modelling confirmed the strongly stabilising influence of the hydrogen-bonding interactions.
These early results have already attracted the interest of other researchers who are keen to see how this will shape future reaction design. ‘I am very impressed by this study,’ comments Manuel van Gemmeren, a catalysis and ligand design researcher at Kiel University in Germany. ‘Their thorough and rational ligand design exploits H-bonding to supplement the weak interactions in the pre-reactive complex and transition state and is validated by well-designed computations. This design logic could be used to address other challenging substrate classes.’
‘The ligands are robust, versatile and easy to access synthetically,’ says Allegra Franchino, a catalysis and ligand design researcher at the University of Durham, UK. ‘The mechanistic insights will pave the way for the extension of the substrate scope and possibly the development of enantioselective versions of this method using prochiral alcohol substrates.’
Yu’s team are aware that this work is still at the proof-of-concept stage but are excited to expand the capabilities of the method. ‘Currently the functional group tolerance is limited and we can only obtain high yields with tertiary alcohols but we anticipate that these limitations will be addressed by further improvements in ligand design,’ comments Strassfeld. ‘The most obvious next steps are to directly expand on these results in terms of the scope of alcohol substrates and the functionalisation step, including other classes of C-C bond forming reactions and C-heteroatom bond formation.’