Delivering a cancer medication to a tumor is merely part of the challenge. After just a few hours of reaching the tumor, numerous therapeutics start to disperse, diluted by blood flow, expelled by pumps located in cancer cell membranes, or simply diffusing into nearby healthy tissue before they can take effect. In other words, the tumor does not retain them. This lack of retention, more than any other factor in drug development, could clarify why treatments that seem promising in laboratory settings frequently fail in clinical applications. A group at the University of California, San Francisco has invested years attempting to resolve this issue, and their most recent findings point to a rather refined solution: a molecular grappling hook that attaches physically to the cancer cell membrane and does not release its grip.
The mechanism, detailed in a paper published this week in ACS Central Science, operates by taking advantage of one of the tumor’s own biochemical peculiarities to secure drugs in position rather than allowing them to disperses.
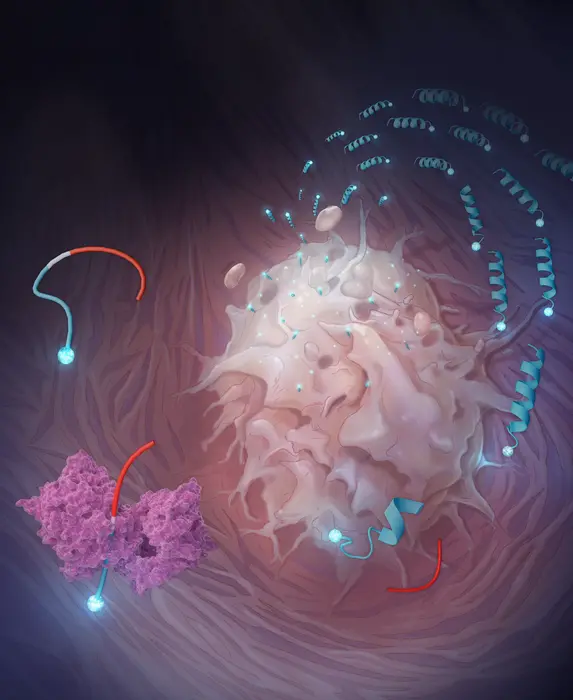
Central to this strategy is a category of engineered peptides that the researchers refer to as restricted interaction peptides, or RIPs. Each peptide is approximately 30 amino acids in length and constructed in three segments, similar to a Swiss Army knife that has been closed. The first segment, derived from an antimicrobial peptide known as Temporin L, serves an active purpose: it can assume a corkscrew-like configuration and penetrate cell membranes. The second segment functions as a masking domain that keeps the first segment tucked away and inactive, prevented from interacting with anything due to its electrical charge. Positioned between them is the crucial component: a short sequence specifically tailored to be cleaved by a protease enzyme that the tumor generates in high amounts. Keep the peptide intact, and the masking domain performs its function, keeping the entire assembly dormant. Cut it in the appropriate spot, and the membrane-binding end is released.
The relevant protease is fibroblast activation protein, or FAP. This serine protease is expressed at elevated levels by cancer-associated fibroblasts, which are supportive cells integrated within the stroma of solid tumors, present in nearly all cancer types. FAP has long been of interest as a therapeutic target, partly because of its consistent expression across types and its significant absence from normal adult tissues. Initial attempts to inhibit it pharmacologically encountered difficulties (protease redundancy indicates that inhibiting one enzyme typically yields minimal effects), but the innovative approach takes the opposite route: rather than blocking FAP, it utilizes FAP as an activator.
Engineered for Adhesion
To create the FAP-activated variant, termed FRIP, the team first needed to identify the precise amino acid sequences that FAP favors for cleavage. They evaluated 228 peptides concurrently employing a method known as multiplex substrate profiling via mass spectrometry, effectively conducting hundreds of cleavage reactions at once and monitoring the outcomes by mass. The standout was a sequence denoted HIGP-TAAY, which FAP cleaved at a rate approximately similar to, and in certain respects quicker than, collagen, which is FAP’s natural substrate in tissue.
What transpired next, when FRIP encountered a live cancer cell, was observable in real time using a confocal microscope. When fluorescently tagged FRIP was added to U-251 glioblastoma cells (chosen due to their high FAP expression), it formed a distinct ring of fluorescence around the cell membrane within approximately 10 minutes. By the 20-minute mark, the signal had migrated inward: the peptide, along with whatever it was transporting, had been entirely internalized. If FAP is blocked with an inhibitor first, the ring fails to materialize. Using a cell line with low FAP expression results in a sluggish effect. The mechanism is about as straightforward as it can get.
For the drug delivery evaluation, the group conjugated MMAE (monomethyl auristatin E), a powerful cell-killing compound widely utilized in antibody-drug conjugates. The combination proved lethal to glioblastoma cells at concentrations roughly 30 times lower than those required for the unbound drug, indicating that the membrane-tethering step was delivering significantly more payload into cells than passive diffusion could achieve on its own. Michael Evans, one of the study’s corresponding authors, has asserted that holding drugs within tumors is possibly the most neglected aspect of cancer pharmacology. The data supports his assertion.
Outsmarting the Clinic’s Benchmark
In mice with head and neck cancer tumors, FRIP loaded with MMAE effectively reduced tumor size more than the drug alone did, and without the body weight loss that led to the free-drug group being withdrawn from the study by day 8. This particular detail is significant: MMAE can be quite toxic at the doses necessary to achieve therapeutic effects systemically. Attaching it to a tumor-specific delivery system seems to considerably mitigate the collateral damage, allowing the drug to concentrate where it is most needed without causing adverse effects across the rest of the body.