Within a deteriorating brain cell, the first component to disintegrate is the nucleus. Not the entire cell simultaneously, nor any neat programmed shutdown, but the nucleus itself: the membrane enclosing it distorts, the smooth sphere becomes wrinkled and shriveled like fruit that has lingered too long in a bowl, ultimately disintegrating. Scientists at King’s College London have coined a term for this. They refer to it as karyoptosis, and they believe it explains a significant portion of neuronal loss that has been vaguely attributed to dementia for quite some time.
This term, karyoptosis, has been circulating in the laboratory for ten years. What is novel is the recent discovery of its presence.
For an extended period, the prevailing understanding resembled this: In Alzheimer’s disease, frontotemporal dementia (FTD), and motor neuron disease, toxic proteins accumulate within neurons until they cease functioning. That seems clear. But how does this occur exactly? Apoptosis, the systematic self-destruction mechanism cells engage in when damaged or unnecessary, was the clear frontrunner, taking most of the blame for years. The issue is that apoptosis didn’t quite account for everything. Mature neurons exhibit remarkable resistance to it, and when you tally up all the apoptotic deaths, a stubborn excess of dead cells remains that passed away through alternative means.
Karyoptosis, the researchers at King’s argue, constitutes at least part of that alternative means.
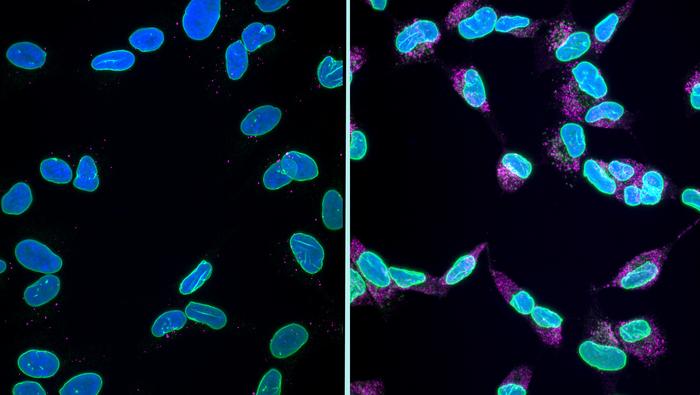
The study, published in Nature Communications, commenced with cultured cells. When the scientists obstructed the cell’s protein degradation system (the autophagy-lysosome pathway, acting as a sort of cellular recycling center), proteins accumulated and the nuclear lamina began to disintegrate. The lamina is a protein network lining the nucleus’ interior, maintaining its shape; one crucial element is a protein known as LaminB1. As LaminB1 became destabilized, the nucleus lost its circular shape, reduced in size, and started to release fragments of itself, encapsulated in small bubbles, into the extracellular space. This was followed by DNA damage. Importantly, the genetic injury occurred after the nuclear collapse, not prior to it. This sequencing is significant because it distinguishes karyoptosis from the gradual degradation associated with cellular senescence, which it superficially mimics.
“The death and loss of cells in the brain drives many symptoms experienced by individuals living with dementia,” remarks Rebecca Casterton, a senior researcher at the UK Dementia Research Institute at King’s and the primary author of the paper. “We have begun to outline the roadmap of how karyoptosis functions.”
Identifying the Switch
A roadmap is only valuable if it directs towards something, and in this case, it leads to a protein known as p38 MAP kinase. Kinases act as the cell’s switches, activating or deactivating other proteins by adding phosphate groups to them, and the research team discovered that p38 phosphorylates LaminB1 at a specific site, destabilizing it and pushing the nucleus toward destruction. When they inhibited p38 using a drug, or modified neurons so that LaminB1 could not be phosphorylated at that specific point, the nuclear damage subsided. They observed this rescue effect in rat neurons, then in fruit flies harboring the genetic mutation linked to one inherited variant of ALS and FTD, and finally in human neurons derived from stem cells. In all three models, reducing that single molecular interaction preserved nuclei and prolonged cell viability.
“By precisely targeting the interaction between p38 MAP kinase and LaminB1, we might mitigate the cell death process, allowing more time for targeted therapies against specific neurodegenerative diseases,” states Manolis Fanto, who led the research.
None of this would hold much significance for human patients if karyoptosis remained confined to the lab. Thus, the researchers turned to the brain bank. They collected post-mortem frontal cortex samples from individuals who had died from Alzheimer’s or frontotemporal lobar degeneration, along with samples from similarly aged individuals without these conditions, and processed thousands of individual cells using a machine-learning sorting technique that was unaware of the source of each cell. The blind algorithm identified clusters of cells exhibiting the shriveled, low-circularity nuclei characteristic of karyoptosis. Upon revealing the labels, those clusters were significantly overrepresented in the diseased brains. Approximately 35 percent of cells from the frontal cortex of Alzheimer’s patients exhibited signs of karyoptosis, in contrast to about 15 percent in healthy aged controls.
It’s important to consider that 15 percent in the healthy brains suggests that karyoptosis isn’t solely a disease-related occurrence; some of it appears to be coming