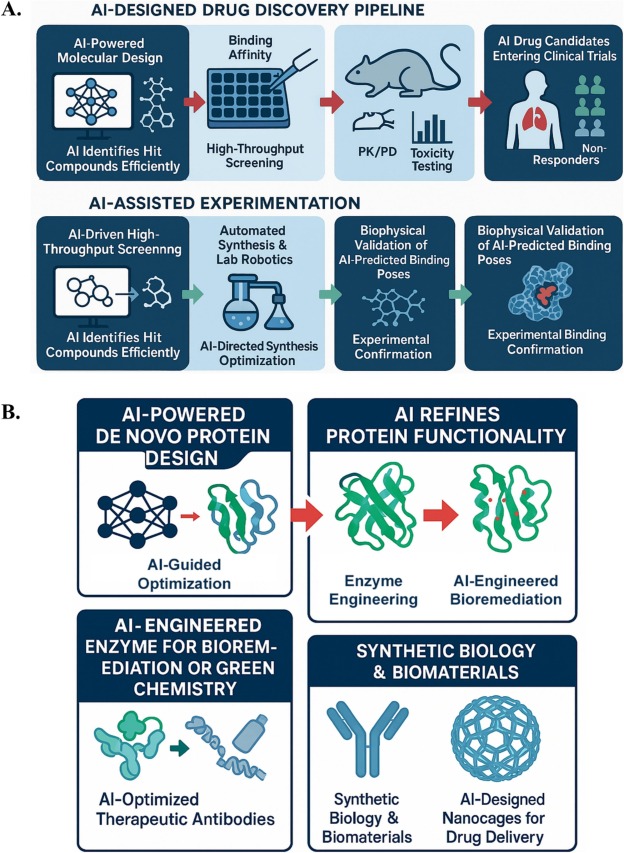
**An Innovative Machine Learning Solution for Drug Development**
Scientists have introduced an innovative machine learning solution aimed at effectively screening small molecules to pinpoint those that may serve as viable candidates for interacting with specific proteins. This state-of-the-art model requires considerably less computational resources than conventional techniques, marking a significant leap forward in the realm of pharmaceutical research. The tool’s ability for high-throughput screening offers the chance to swiftly identify small-molecule ligands for every druggable target found in the human genome, potentially promoting pharmaceutical advancements.
Contemporary methods in artificial intelligence, especially those utilized for predicting and designing protein structures, have proven essential in drug development and were notably awarded the 2024 Nobel Prize in Chemistry. Nevertheless, leveraging such approaches for the discovery of ligands that can interact with proteins is resource-intensive. As per Yinjun Jia from the Institute for AI Industry Research at Tsinghua University, the molecular docking process is inherently slow due to the requirement of evaluating various angles and poses to determine the optimal molecular fit, a practice that remains labor-intensive even with current computing capabilities. This computational bottleneck elucidates why merely around 10% of the 20,000 protein-coding genes within the human genome currently have well-documented small molecule binders.
The newly devised DrugCLIP framework by Jia and his team presents a revolutionary method. This cutting-edge system conceptualizes both protein pockets and potential small-molecule binders as vectors within a high-dimensional space. By computing the scalar product of these vectors, the tool forecasts binding affinities, forming the foundation of a deep-learning algorithm that ranks potential ligands based on their anticipated affinity for target protein pockets. To confirm binding feasibility, the researchers employ AlphaFold3 to ascertain the exact binding conformations of the leading candidates, achieving a screening efficiency up to ten million times quicker than traditional docking processes.
The effectiveness of this approach was demonstrated through the identification of novel molecules targeting crucial psychopharmacology proteins: the serotonin 2A receptor and the norepinephrine transporter. Laboratory evaluations, encompassing biochemical assays and cryo-electron microscopy, validated the efficacy of these molecules. Remarkably, a molecule discovered for the norepinephrine transporter exhibited superior chemical efficacy compared to bupropion, a widely used antidepressant, although Jia stresses the importance of clinical validation. Promisingly, the team has also uncovered another notable molecule, which they plan to progress toward clinical trials.
Nicholas Polizzi from Harvard Medical School voices measured optimism regarding this advancement. He acknowledges the promise of a high-throughput, genome-wide screening technique to uncover ligands that specifically engage proteins with minimized off-target effects, a substantial benefit in mitigating drug adverse effects. However, Polizzi raises a concern: the structural resemblances of protein pockets across various protein families may pose challenges in establishing whether the deep-learning model has genuinely generalized binding principles or merely memorized details from the training data. Despite these apprehensions, Polizzi recognizes the groundbreaking potential of the research, indicating that if pre-screening and molecular docking simulations can realize the anticipated accuracy with diminished computational demands, it signifies a crucial step forward in drug discovery.